醫學遺傳學/酶活性降低引起的遺傳性酶病
| 醫學電子書 >> 《醫學遺傳學基礎》 >> 單基因病 >> 基因突變致酶活性異常 >> 酶活性降低引起的遺傳性酶病 |
| 醫學遺傳學基礎 |
|
|
|
1.酶活性降低的原因基因突變引起酶活性降低的可能原因包括:①結構基因突變;其結果致使酶動力學特性改變(表現為酶與底物親和力降低,與抑制物的親和力增高)和酶的穩定性降低(表現為酶降解速率加快);②調節基因突變:酶合成速率減慢;③影響翻譯后修飾和加工。
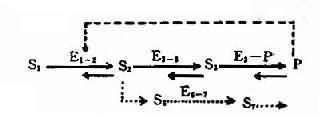
圖表-21 典型代謝途徑示意圖
S1:底物;S2、S3:中間產物; P:終產物;E1-2、E2-3:不同代謝環節的酶;
E3-P:形成終產物的酶;E6-7:旁路代謝的酶;S6、S7:
旁路代謝產物;---旁路代謝途徑;---反饋抑制
2,酶活性降低的發病機理 人類體內一般代射過程如圖4-21所示:代謝底物(S1)進入細胞后,底物(S1)在一系列酶(E+1-2。E2-3,E3-P)催化下經過一系列中間產物(S2、S3),最后形成代謝終產物(P),同時代謝產物(P)可對一定的酶(如E1-2酶)起反饋抑制作用。另外,在正常代謝過程中還可能存在代謝旁路(S2→S6→S7……)。當控制酶蛋白合成的基因發生突變時,正常代謝受阻,可能通過不同發病環節引起不良后果。
(1)酶缺乏致代謝中間產物堆積和排出引起的疾病:當E2-3缺乏時,中間產物S2不能轉變為S3,它在血和尿中的濃度增加,如果中間產物是無毒的,可由腎排出或通過其它方式降解,則不會嚴重危害人體。如尿黑酸尿平患者,因酪氨酸代謝中間產物(尿黑酸)堆積,可大量由尿排出,對人體無影響。但如果中間產物具有毒性,則將會引起癥狀。
半乳糖血癥可作為實例,半乳糖血癥(galactosemia)表現為嬰兒哺乳后嘔吐、腹瀉,對乳類不能耐受,繼而出現肝硬化、白內障、智力發育不全等癥狀。乳類含有乳糖,它經消化道乳糖酶分解產生葡萄糖及半乳糖,半乳糖通過一系列酶促反應產生葡萄糖而被組織利用(圖4-22)。
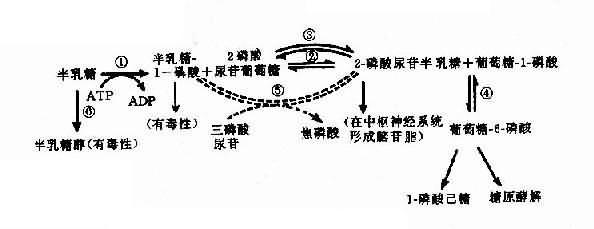
圖4-22 半乳糖代謝途徑(虛線表示年長以后才發展起來的代償途徑)
①半乳糖激酶;②半乳糖-1-磷酸尿苷酰轉移酶;③半乳糖尿苷-2-磷酸-4-異構酶;④磷酸葡萄糖變位酶;⑤-磷酸尿苷半乳糖(或葡萄糖)焦磷酸化酶;⑥醛糖還原酶
典型的半乳糖血癥患者由于半乳糖-1-磷酸尿苷酰轉移酶(galactose-1-phosphate uridy1 transferase,簡稱轉移酶)缺乏(圖4-22②),致使半乳糖-1-磷酸(Gal-1-P)及半乳糖積聚在血中,部分隨尿排出。Gal-1-P在肝的積聚可引起肝功能損害,甚至肝硬化;在腦的積聚引起智力障礙;血中半乳糖升高可使葡萄糖釋出減少,出現低血糖癥。半乳糖在醛糖還原酶作用下產生半乳糖,能改變晶狀的滲透壓,使水分進入,影響晶狀體代謝而致白內障。
患者都是隱性純合子(gg),雜合子表型正常,轉移酶活性約為50%,活性低于10%可出現典型癥狀。
另一類半乳糖血癥為半乳糖激酶(galactokinase)缺乏所引起,癥狀較輕,主要表現為青年型白內障,血中半乳糖增高,但無肝及腦損害。
轉移酶基因(GALL)定位于9p13,半乳糖激酶基因(GALK)定位于17q21-q22。兩病均為常染色體隱性遺傳。
(2)酶缺乏致代謝底物堆積引起的疾病:當一系列生化反應可逆時,一處的阻斷常導致代謝底物(S1)貯積。貯積的物質如果溶解度高,則在血和尿中濃度增高;若溶解度低時,則在組織中貯積引起疾病。
糖原貯積癥(glycogenstorage liseaee,GSD)是一組由糖原合成和降解酶缺陷引起的疾病,至少有12種類型。糖原貯積癥主要累及肝或肌肉,但有的也可伴有心、腎和神經系統的損害。不同類型之間其嚴重性和預后都不全相同。例如,von Girke病(Ⅰ型)癥狀非常嚴重,而Hers病(Ⅵ型)較輕。此類疾病發病機理可用von Gierke病為例說明。本病是由于肝內葡萄糖6-磷酸酶(glucose-6-phosphatase,G6Pase)缺乏引起。肝糖原在一系列酶的作用下生成葡萄糖,這個反應的各個步驟都是可逆的,其主要步驟如下:
![]()
患者由于G6Pase缺乏,所以G6P不能轉變為葡萄糖供組織利用,通過可逆反應而合成過多的肝糖原,引起患兒肝腫大。當不進食時極易發生低血糖。由于動用脂肪可以出現酮血癥。G6P通過無氧酵解,生成大量乳酸,導致酸中毒。所以患者的肝大伴低血糖,發育不良,消瘦,身體矮小,常有出血傾向。肝活檢見糖原含量增加。
本病呈染色體隱性遺傳。患者G6P酶完全缺乏,多數患者父母表型正常,但G6P酶活性為中間值。同胞中可出現患者。
(3)酶缺乏致代謝終產物缺乏引起的疾病:這是一類由E2-3酶缺乏,致所有產物(包括終產物P)缺乏引起的疾病.白化病就是實例.白化病(albinism)分全身型及局部型.全身型常見,患者皮膚呈白色,毛發銀白或淡黃色,虹膜及瞳孔呈淡紅色,視網膜無色素,羞明,眼球震顫等,本病發病率約1/10000-1/20000,呈常染色體隱性遺傳.
正常人黑色素由黑素細胞合成,這些細胞中有特殊的細胞器-黑素小體(melanosome),其中有含銅的酚氧化酶,即酪氨酸酶(tyrosinase),它可將酪氨酸轉變成黑色素(圖4-23).白化病人有黑素細胞,但酪氨酸酶缺乏,使黑色素不能形成,病人因缺黑色素而白化.白化病存在遺傳異質性.已知白化病至少有7種不同類型.酪氨酸酶基因(TYR)定位于11q14-q22.
屬于此類的例子尚有遺傳性甲狀腺腫,它是由于偶聯酶、脫碘酶或碘過氧化酶缺乏導致甲狀腺素生成減少引起的,患者身材矮小,智力低下,面貌丑陋.
(4)酶缺乏致旁路產物增多引起的疾病:當酶的缺乏導致主要代謝途徑受阻斷時,過量的前體物S2通過另一旁路代謝引起某些副產物的堆積.如果增多的旁路產物是無毒的,則可排出體外不致危害人體,但如果旁路產物或其分解產物有毒,則可危害機體引起疾病.
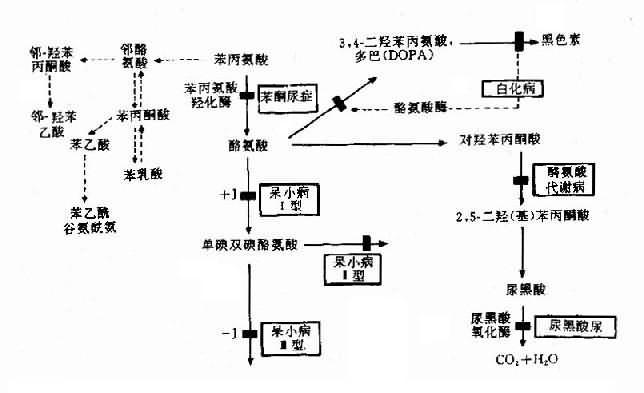
圖4-23 苯現氨酸代謝及有關的遺傳性酶病的發病機理
苯酮尿癥(phenylketouria,PKU)可作為實例。本病以智能發育不全為主要特征,呈常染色體隱性遺傳,群體發病率約1/16000,由苯丙氨酸羥化酶(phenylalaninehydroxylase,PAH)遺傳性缺乏引起。現已知苯丙氨酸羥化酶基因定位于12q24.1,此基因全長約90kb,含13個外顯子,在中國人中已發現10余種點突變,這是造成酶活性缺乏的原因。
典型PKU患兒出生時,外貌正常,約至3-4個月時,漸出現智能發育不全,患兒步伐小,姿似猿猴,肌張力增高,易激動,甚至驚厥,90%以上患者毛發發黃,膚白,甚至虹膜呈黃色(白種人呈藍色)。此外,患兒尿和汗有一種特殊的腐臭。
正常人苯丙氨酸在體內主要通過PAH的作用轉變成為酪氨酸,繼而生成黑色素(圖4-23)。PAH主要存在于肝中,它的作用需要輔因子四氫生物蝶啶(BH4),如果PAH缺乏(活性<1/10)可阻斷苯丙氨酸轉化為酪氨酸,即產生典型的苯酮尿癥。酶活性部分缺乏,可導致輕度高苯丙氨酸血癥(hyperphenylalaninemia)。
典型PKU患者,由于肝內PAH幾乎完全缺如,致血清苯丙氨酸增高(正常人1-3mg%,患者可達50-100mg%)。過多的苯丙氨酸通過轉氨酶作用生成苯丙酮酸,再經氧化.脫羧產生苯乳酸.苯乙酸等異常產物,由尿和汗液排出,致患兒的尿和汗呈特殊的腐臭。可能由于旁路產物抑制了腦組織內L-谷氨酸羧酶,使谷氨酸脫羧基生成γ-氨基丁酸減少,而后者對腦細胞的發育及功能起重要作用。也有人認為苯現氨酸和旁路產物可影響5-羥色胺的生成,而影響腦功能。由于正常產物酪氨酸是黑色素前體,所以酪酸不足加之旁路產物可以抑制酷氨酸酶,使患者皮膚毛發及眼血管膜色素減少,因而色澤都較淺。
(5)酶缺乏致反饋抑制減弱引起的疾病:有些代謝過程中,某些代謝產物對整個反應過程具有反饋調節作用。因此,某種酶的遺傳性缺陷,使該代謝產物減少,致反饋調節功能失調。自毀容貌綜合征可作為實例。處毀容貌綜合征亦稱Lesch-Nyhan綜合征(Lesch-Nyhan syndrome,LNS),是由于遺傳性次黃嘌呤鳥嘌呤磷酸核糖轉移酶(hypoxanthine-guanine-phosphoribosyltransferase,HGPRT)缺乏所引起。本病的特征是智力發育不全、舞蹈樣動作和強迫性自殘行為,并伴有高尿酸血癥、尿酸尿、血尿、尿道結石和痛風。患者可活至20余歲,多死于感染和腎功能衰竭。
LNS呈X連鎖隱性遺傳,患者均為男性(半合子)。現知HGPRT定位于Xp26-q27.2,其發病率約為1/380000。
正常時,HGPRT的功能是將次黃嘌呤轉變為次黃苷酸(((即IMP,肌苷酸),將鳥嘌呤轉為鳥苷酸(GMP),鳥苷酸及腺苷酸可以反饋抑制磷酸核糖焦磷酸(PRRR)合成1-氨基-5-磷酸核糖,從而控制IMP的自發合成速度(圖4-24)。這一過程,HGPRT的催化起到反饋抑制作用。當HGPRT缺乏時,這一反饋抑制作用減弱或消失,IMP和GMP生成量大為減少,嘌呤合成速度就大大增快,致使患者體液內的尿酸濃度增高,出現高尿酸血癥和尿酸尿。經典型患者無HGPRT活性。酶僅部分缺乏時,患者往往出現痛風而無LNS癥狀。
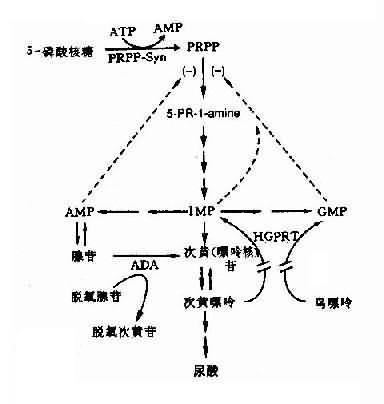
圖4-24 HGPRT缺乏導致Lesch-Nyhan綜合征
5-PR-1-amine:-1氨基-5-磷酸核糖;PRPP-Sym:磷酸核糖焦磷酸全盛酶;虛線:反饋抑制
(6)維生素依賴性遺傳病:這是由于基因突變改變了某種酶蛋白,使之與輔酶的相互作用受到損害引起該酶活性降低。這類輔酶多數為維生素,故稱為維生素反應性遺傳病(vitam-in-responsive hereditary disorders)。例如,在體內,吡多醇-5-磷酸(PLP)是維生素B6的活性形式,它是多種酶蛋白的一種輔酶,因此,某些酶蛋白基因的突變能引起多種遺傳性代謝病(圖4-25)。例如谷氨酸脫羧酶基因的突變致使酶蛋白和PLP的相互作用受損,將導致γ-氨基丁酸缺乏和痙攣。胱硫醚合成酶基因突變使酶蛋白與PLP的結合部位發生改變而導致胱硫醚尿癥。胱硫醚合成酶的缺乏可導致同型胱氨酸尿癥。
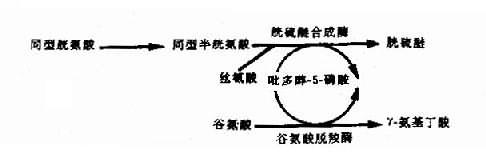
圖4-25 吡多醇-5-磷酸(PLP)作為多種酶的輔酶形式示意圖
(7)多種酶缺陷引起的疾病:有些遺傳性代謝缺陷,在某一患者不只一種酶缺陷。例如先天性蔗糖不耐受癥(congenital sucrose intolerance),患者其異麥芽糖酶和蔗糖酶都缺乏,而楓糖尿癥(maple syrup urine disease)病人體內則同時缺乏纈氨酸脫羧酶.亮氨酸脫羧酥酶和異亮氨酸脫羧酶。這種現象可作如下解釋:①有缺陷的幾種酶均有一條共用的多肽鏈,當編碼這條多肽鏈的結構基因發生突變時,就會使凡含有這條共同多肽鏈的各種酶蛋白結構改變而失活;②由于一個酶,致使由這種催化生成的代謝物缺乏,因此由它誘導的各種酶也相應缺乏;③由于一個調控基因發生突變,關閉了鄰近幾個結構基因,使這些結構基因控制的酶不能產生。
| 關于“醫學遺傳學/酶活性降低引起的遺傳性酶病”的留言: | |
|
目前暫無留言 | |
| 添加留言 | |